I think there more work needed to have a fair and clear description of the importance to limitations to genomic analysis, but I believe concepts like over-fitting and variability also be important.
Here is a plot that I believe can help explain what I mean by over-fitting :
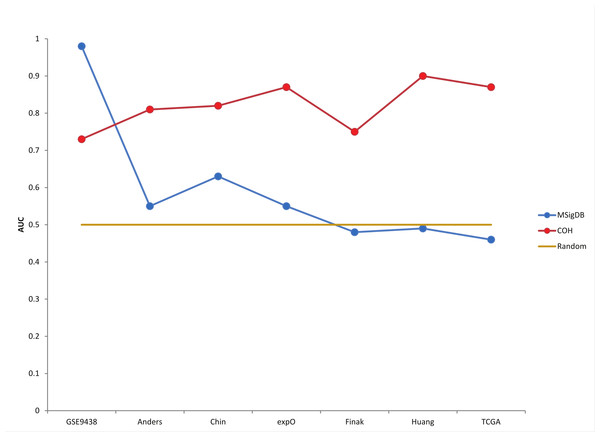
The plot above is from Warden et al. 2013. Notice that very high accuracy in one dataset (from which a signature was defined) actually resulted in lower accuracy in other datasets (which is what I mean by "over-fitting"). The yellow horizontal line is mean to be like the diagonal line in AUC plots. While 50% accuracy is not actually the baseline for all comparisons (for example, if an event or problem is rare, then saying something works 0% of the time or 100% of the time may actually be a better baseline). Nevertheless, I think this picture can be useful.
When I briefly mentioned "variability," I think that is what is more commonly thought of for personalized / precision medicine (solutions that work in one situation may not work as well as others). However, I also hope to eventually have an RNA-Seq paper to show that testing of multiple open-source programs can help towards having an acceptable solution (even if you can't determine the exact methods should be used for a given project ahead of time). I think this is a slightly different point in that it indicates limits to precision / accuracy for certain genomics methods, while still showing overall effectiveness in helping answer biological questions (even though you may need to plan to take more time to critically assess your results). Also, in the interests of avoiding needless alarm, I would often recommend people/scientists visualize their alignments in IGV (a free genome browser), along with visualizing an independently calculated expression value (such as log2(FPKM+0.1), calculated using R-base functions); so, if you think a gene is likely to be involved, that can sometimes be a way to help gauge whether the statistical analysis produced either a false positive or a false negative for that gene (and then possibly provide ideas of how to refine analysis for your specific project).
This is also a little different than my earlier blog post about predictive models in that I am saying that over-fit models may be reported to be more accurate than they really are (whereas the the predictive power of the associations described in that post clearly indicate population differences have limitations in predictive power in individuals). However, I think that level of predictive power for the SNP association is in some ways comparable to the "COH" gene expression model shown above (where roughly 80% accuracy is actually more robust, and therefore arguably more helpful, than a signature with >90% accuracy in one cohort but essentially random predictions in most other cohorts).
I think that also matches this Wu et al. 2021 commentary, where performance noticeably drops in Table 1 when an AI model is trained at one site and tested at another (with highest performance coming from the same site as training). However, this is a little different than what I showed above (where lower performance on the same dataset may result in relatively better performance in different datasets for the BD-Func plot, but I think the maximal performance is more correlated in the AI commentary table with a loss across in performance in new sites across all 3 rows). If there was at least 1 additional row with non-AI models that had more similar performance on the same site and different sites (but lower performance than the AI model on the same site), then that would be more similar to the BD-Func example.
Also, I think it should be emphasized that precision medicine doesn't necessarily have to involve high-throughput sequencing, and I think using RNA-Seq for discovery and lower-throughput assays in the clinic is often a good idea. For example, the goal of the paper was a little different, but the feature being predicted in that plot above is Progesterone Receptor immunostaining (I believe protein expression for ER, PR, and HER2 are often checked together for breast cancer patients). So, just looking at the PGR mRNA might have had more robust predictions in validation sets than the BD-Func "COH" score (which was a t-test between up- an down-regulated gene expression, per-sample).
There are positive outcomes from genomics research, and there are some things that can be known/performed with relatively greater confidence than others (such as well-established single-gene disorders). However, I think having realistic expectations is also important, and that is why I believe there should be emphasis on both "precision medicine" and "hypothesis generation" when discussing genomics. Or, I actually prefer the term "personalized medicine" over "precision medicine," which I think can capture both of those concepts.
Change Log:
5/11/2019 - this tweet had some influence on re-arranging my draft (prior to public posting), in terms of the expectation that personalized medicine / genetics can explain / improve therapies that originally did not seem very effective.
5/18/2019 - public blog post
5/20/2019 - update link for "personalized medicine," add sentence in 1st paragraph, and remove "medicine" from title (and one sentence in concluding paragraph).
5/22/2019 - I don't remember why I had this in the draft for the generics post. While I don't think it fits in with the flow of the main content, I wanted to add this as a slide note relevant to general limitations in precision (even when a program is incredibly useful): As mentioned in this tweet, BLAST is of huge benefit to the bioinformatics / genomics community, even without choosing a "typical" 0.05 E-value cutoff (to be more like a p-value).
5/26/2019 - add Mendelian Disease as "success story" for genomics
4/8/2021 - add link to article about issue with retrospective studies and decrease in performance between sites




No comments:
Post a Comment