1) Increased false positives for DNA-Seq variants at lower frequencies in normal controls
From my own publication record, I discuss this in Warden et al. 2014:
Namely, Figure 7 shows an implausibly high proportion of damaging variants with lower-frequency germline variants using VarScan with default settings (in healthy 1000 Genomes controls):
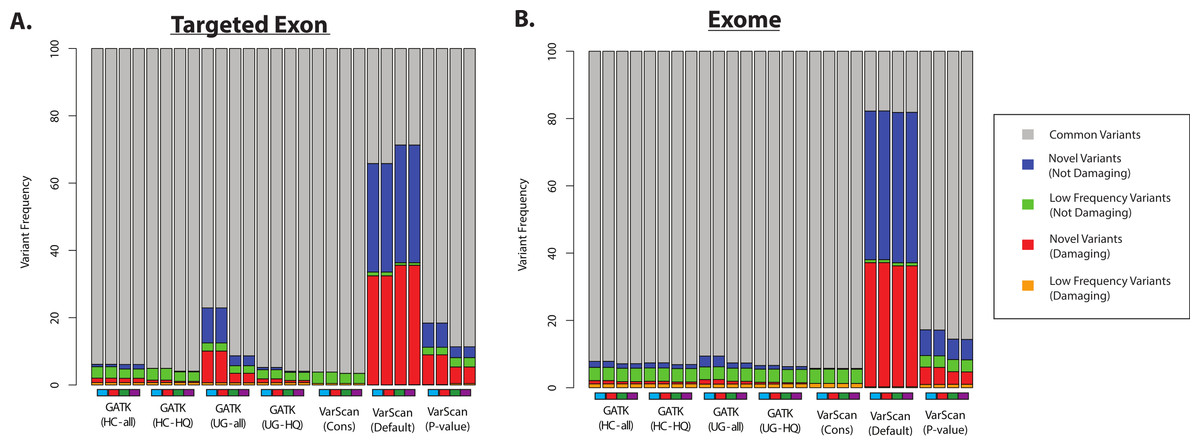
The variant frequencies (per sample, at a given variant position) are not immediately obvious from the above plot. However, you can see those "novel" variants are more likely to be detected with the default settings in VarScan (Figure 4 of the Warden et al. paper):
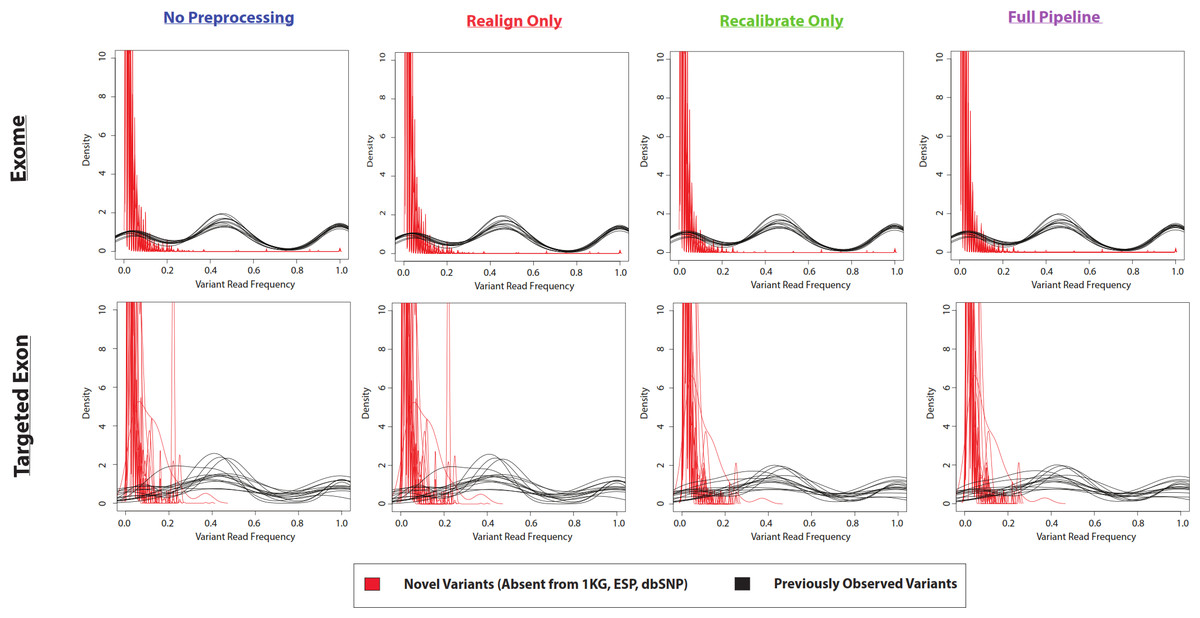
Although, importantly for this discussion, those variant calls can be improved with filtering. Notice the "novel" and "known" variant frequencies are more similar, which is one of the indications in that paper of a lower false positive rate (Figure 5 of the Warden et al. paper):
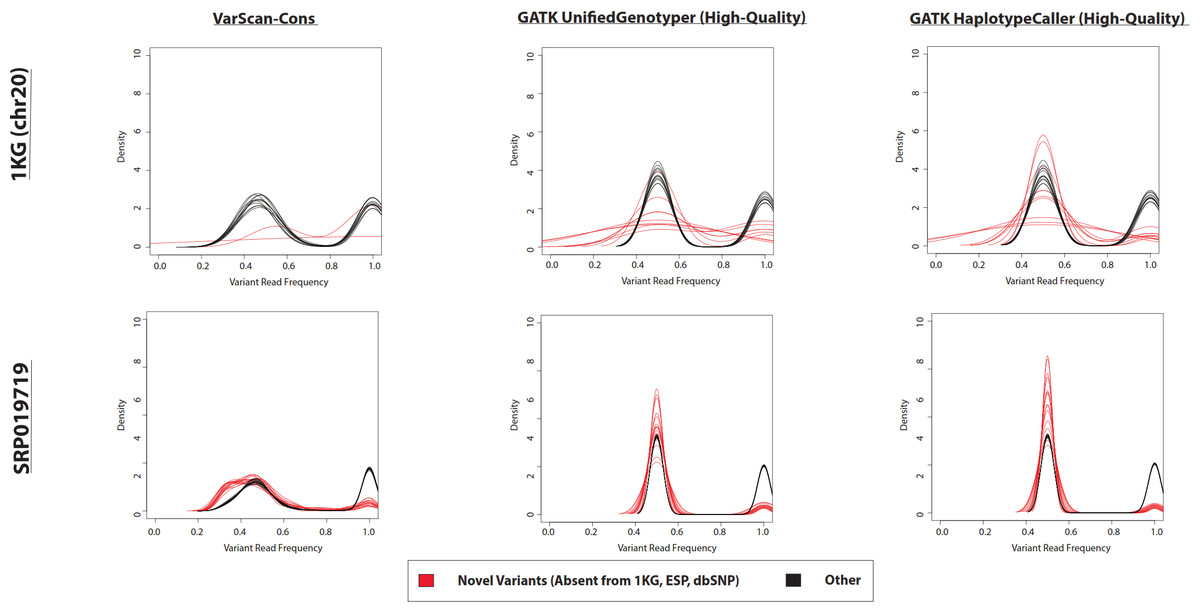
In terms of other publications, you can also see increased discrepancies for low-frequency somatic variants (less than 20% variant fraction) in Arora et al. 2019 pre-print (in Figures 5B and 5D), as well as Figure S1 of Yizak et al. 2019 (although the emphasis on RNA-Seq versus DNA-Seq data is a little different).
2) Possible barcoding issues, such as with PhiX sequence (which doesn't have a barcode and theoretically shouldn't be in any de-multiplexed samples, even though you usually see at least some PhiX reads).
For 2), I would be happy if this post got enough attention for people to be aware there is a non-trivial chance their top BLAST hit for a PhiX sequence could be incorrectly labeled as a 16S sequence.
While a little less clear, I also think there should be more post-publication review for this eDNA article, where I think the title is the opposite of what seems like the most parsimonious explanation.
I added this quite a bit after the original post. However, to be fair, you may sometimes be able to get some idea about barcode hopping if you have a small fragment (where you actually sequence the barcodes past your genomic sequence). This is what I did for my cat's basepaws sequence (~15X WGS): in that case, you could see some other valid Illumina barcode combinations among my reads, but it wasn't too bad. I'm not sure if this could be more of an issue with the low-coverage sequence (less than 1x), but I have general concerns about that (for most applications, except broad ancestry or IBD calculations).
Update Log:
6/9/2019 - original public post
8/2/2019 - add link to basepaws script
8/9/2019 - replace GitHub link with blog post link
8/2/2019 - add link to basepaws script
8/9/2019 - replace GitHub link with blog post link




This comment has been removed by a blog administrator.
ReplyDeleteThis comment has been removed by a blog administrator.
ReplyDeleteThis comment has been removed by a blog administrator.
ReplyDeletePretty good post. I just stumbled upon your blog and wanted to say that I have really enjoyed reading your blog posts. Any way I'll be subscribing to your feed and I hope you post again soon. Big thanks for the useful info.
ReplyDeleteCurso Ciencia de Datos
This comment has been removed by a blog administrator.
ReplyDeleteThis comment has been removed by a blog administrator.
ReplyDeleteThis comment has been removed by a blog administrator.
ReplyDeleteThis comment has been removed by a blog administrator.
ReplyDeleteThis comment has been removed by a blog administrator.
ReplyDeleteThis comment has been removed by a blog administrator.
ReplyDeleteThis comment has been removed by a blog administrator.
ReplyDelete