Part #1: Short Summary:
In the basepaws Facebook group, there are some noticeable issues with results for purebred cats that are not already part of the reference set (as I understand it). To be fair, I have seen at least one report that was a good result (matching the known breed in a new test cat), but I think independent test datasets tend to be discordant with known purebred cats more often than they are concordant with new purebred cats (in the small fraction of customers where that can be tested). Also, the majority of cats are mixed breed "Polycats" where there probably shouldn't be any close purebred relatives, and I think the results more-or-less match that expectation (if interpreted correctly, and I have found explaining precisely what I think is "correct" interpretation to be bit of a challenge).
Again, to be fair, basepaws does say "Disclaimer: The Basepaws Breed Index is not a breed test". However, I think there is still some noticeable confusion. So, regardless of whether my suggestion is helpful, I think it is safe to say you should not be using basepaws to confirm your cat's breed or disprove your cat's breed, and you may not have closely related cats for the "breed" with the highest estimated breed fraction in chromosome painting (which, in the wording of the current reports, may be "Breed Group" instead of "Breed Index" - either way, this is what you receive when you order the Breed + Health DNA Test, and you can see some type of chromosome painting in the various reports for Bastu).
After having several very helpful discussions, I thought of a possible suggestion that I thought might help: perhaps use a more abstract name for an ancestry group / cluster to make clear that you shouldn't use what is being provided to define breeds.
I think the best example may be the broad "Eastern" ancestry group, which is already in the reports (as a broader category) as well in previous literature on cat genomics.
If calculated accurately, I would expect Siamese cats will tend to have a higher estimate of Eastern contribution (at least for the "Traditional" Siamese cats, as I understand it). However, I think the Colorpoint Restriction mutation might be what owners might want to check for cats with the characteristic hair color pattern. For example, you might have to dig around a bit in the comments for this video but I believe "Lynx Point" cats can be defined from 2 markers (the Colorpoint Restriction variants, and Agouti variants for the tabby pattern).
Nevertheless, my point is that I believe giving the ancestry group the "Eastern" name adds a layer of conceptually separation than the "Siamese" (or related) breeds.
So, if technical problems are minimal, then I think this might reduce confusion for purebred cat owners (as well as minimize miscommunication from misunderstanding). It might also reduce interest, but I would rather plan to have fewer customers than have more customers whose satisfaction might decrease over time.
It might also be worth noting that I believe most imputed genotypes are phenotypically neutral, meaning that they do not cause the traits that are used to define the breeds (although relatedness does define the breed, once it becomes established).
There are some additional details that I will discuss below, but I hope this can help for discussions (instead of me posting several comments, which may or may not be appreciated by all basepaws customers). I also have a "Change Log" since I hope that I can update this based upon feedback, in order to try and be able to explain my point as precisely as possible.
Part #2: Intermediate-Level Details:
I have a separate blog post describing results (from multiple organizations / companies) for my cat Bastu.
However, I will copy / repeat some points below. Some hopefully complement the main point for the "Short Summary," but some of these are separate points.
First, I think seeing 3 technical replicates for my 1 cat may be helpful in giving a sense of confidence in the results:
"Confident" Threshold

"Possible" Threshold

If you look at the broad ancestry groups, then there is some noticeable consistency with the technical replicates. A mostly "Western" contribution for Bastu also makes sense, and that is also consistent with the her Ancestry Test from UC-Davis.
Please also note that the 1st 2 samples (left and middle) are low coverage Whole Genome Sequencing (lcWGS) and the report on the right is "regular" coverage sequencing (~15x).
For example, initiatively, I would not expect Bastu to be related to any Exotic breeds. So, in addition to the general issue that accurately genotyped variation can exist before breed creation and confound the results, you can see the "Exotic" contributions go away in the higher/regular coverage sequencing data.
The main reason that I purchased the "Whole Genome Sequencing" kit was that I would get access to the raw data. Since the price is different now, I will include the analysis part of this file below (without the header listing the original price):
Again, noticed the relatively higher Western contribution is also consistent with my re-analysis.
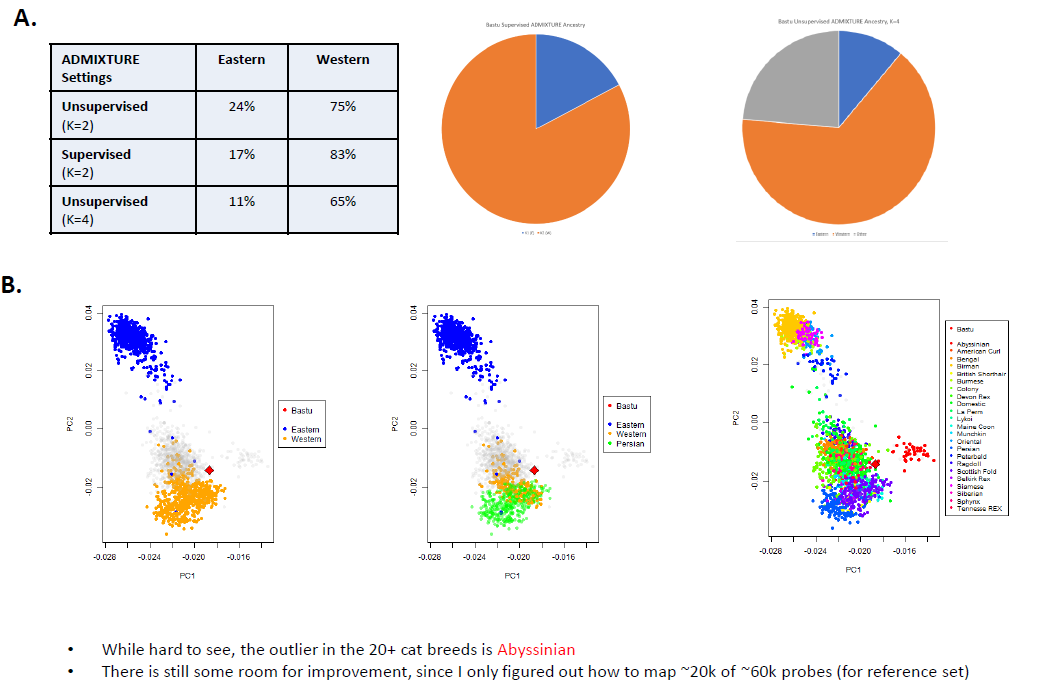
While I want to make clear that the PCA plots (the dots in the bottom part of the image, which I tried to describe as part "B") come from a relatively limited number of SNPs (from public cat SNP chips). So, I don't know what the spread looks like if you could look like if you could see the individual points in the basepaws data, but I think plotting a centroid may not be the best way to represent the data (if that is what was done here).
Part #3: Advanced Details:
With my ~15x cat genome data (along with my human data), I tested running imputation of down-sampled reads, which you can see here. I am also plotting that below:
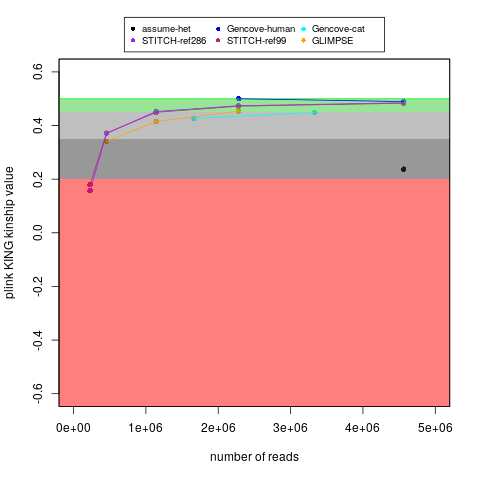
For reference, I don't have the raw lcWGS FASTQ data, but earlier versions of reports said that technical replicate #1 had 3,488,515 fragments and technical replicate #2 had 2,712,098 fragments. For comparison, my regular coverage WGS sample had 166,490,724 paired-end reads (I think comparable to the "fragments" in those reports).
Because of the fragments smaller than the read length, my "regular" coverage WGS was closer to 15x than 20x. I also don't know how many fragments are for lcWGS versus Amplicon-Seq in the "Breed + Health" results. Nevertheless, I think the lcWGS "fragments" in those earlier reports roughly match an expectation of being 0.1x to 0.5x.
My main goal in the above plot was to test self identification. With the same number of reads, the Gencove cat imputation worked a little worse for my cat's sample than my human sample (for either Gencove or open-source options for humans), but I don't think that is surprising because I think the human variation is better understood. Nevertheless, if you follow the Gencove-cat trajectory, then I think there might be some benefits to having ~2x the fragment count (even though I would probably still not consider that to be equivalent to genotypes without imputation). That said, to be fair, I don't know if some things (like broad ancestry groups) might be detectable with less precision than being able to self identify your own (cat's) sample. Also, if a relative finder function is added, then Bastu's 2 lcWGS technical replicates can act as a positive control (to gauge how important this measure really was).
Likewise, I believe there is at least the Martin et al. 2021 paper emphasizing a noticeable advantage to using 4-6x WGS (over 0.5x WGS). For example, there is a noticeable difference for the 0.5x results in Figure 4A (and as well as Table S4 and Table S5). That is for certain populations, but it seems like that may match what I am seeing as well?
Perhaps most importantly, to give some more examples of selected problems on the human side, you can also see issues with the human lcWGS imputation here at the level of individual genotypes, which I think may be why Nebula now only provides regular/high coverage sequencing for customers.
basepaws uses Amplicon-Seq (not the lcWGS) for the health markers, so I believe those should be thought of as independent results (and the technical replicate discordance is not necessarily predictive of the concordance for the health markers).
My understanding is that basepaws is planning on adding Amplicon-Seq for trait markers in the future. I think this will be added to the existing report for the "regular" coverage sequencing data. However, customers will need to keep in mind this can't (or at least shouldn't) be added from the lcWGS. My understanding is that the additional cost to add those extra Amplicon-Seq markers to previous reports will be minimal, but I think that is worth making clear new libraries need to be prepared to return those results.
It might not be the most important point, but I wanted to acknowledge that I believe what basepaws is doing with the chromosome painting is better than produced got for Bastu's sample with RFMix (which you can see here, for example, if you scroll towards the middle of the post). However, I thought that was sufficiently far from being a reasonable result that I did not include that in my mini-report of re-analysis that I showed above. I am also expressing similar concerns for the technical replicates above.
So, in the immediate future, I am not sure if doing something like the ADMIXTURE analysis used for the table and pie charts might help with giving robust results for those broad ancestry groups (which I think should also work with closely related cats, if you used something like plink kinship). If there were any issued with defining relatedness with the chromosome painting results, then I think that that should be OK with enough reads for 1 genome-wide estimate (and I would rather have a close cat relative finder than a breed index/group that might cause confusion and may not be great at identifying breeds in new test samples, like what I have seen in the Facebook discussion group). However, either way, that is the explanation for what I presented in my "mini-report" from re-analysis of raw "regular" coverage sequencing data.
Part #4: Acknowledgements:
I can't thank the other basepaws customers enough for their discussions, which have helped me sort through my thoughts. I also hope that can continue in the future!
The basepaws staff has also provided responses to all of my e-mails. For example, I think that is how I know the Breed Index/Group (which shouldn't be used to categorize purebred cats) comes from lcWGS and the health markers come from Amplicon-Seq (also verified here). So, thank you for your helpful feedback, as well as encouraging a community were such discussion can take place!
P.S. I have some on-going notes about known traits (not currently in the basepaws report), but I would be happy to hear more. This includes things like long hair.
In general, I tend to provide things like links to informational details from the UC-Davis VGL, but I am encouraging you to us the links on the informational page to purchase those separate tests. For example, I purchased the UC-Davis "Cat Ancestry" panel that includes some trait markers as well the Optimal Selection panel (also mentioned in the earlier blog post).
If you are a breeder, I think part of the goal of Optimal Selection is to identify mating pairs with the maximal possible diversity within the breed.
Maybe it is otherwise a little off-topic, but I mention this because I think people purchasing the Breed + Health kit might in fact be interested in understanding some of the traits that they see in thier cat.
P.P.S. If a relative finder was added and both parents were in the basepaws database, then that would in fact be relevant for confirming that a cat is purebred (as long as there were no methodological issues, or especially low read coverage). However, that is not currently offered from basepaws, so that is why I am saying that the current breed measures cannot be used to determine your cat's breed (as also said in the disclaimer from basepaws).
Change Log:
2/27/2021 - public post
2/28/2021 - minor changes (including switching wording from "Breed Index" to "Breed Group")
3/6/2021 - add notes for some other common questions and caveats + minor change
3/15/2021 - add lcWGS fragment counts (and linked image)
3/26/2021 - add note to make clear that all 3 reports were re-processed at the same time + add link to Martin et al. 2021 lcWGS paper
3/28/2021 - add extra details / minor re-organization; correct misunderstanding of table
4/18/2021 - add sentence about basepaws sample mix-up
11/21/2021 - add note about 2 markers defining "Lynx Point".




No comments:
Post a Comment