I already had an earlier Whole Genome Sequencing test from Basepaws, along with other results that can be viewed here or here.
However, I decided to order another test for the following reasons:
- I would like to have some more raw data in order to help be better evaluate what I think of the Dental Health Results
- 15x is a bit low, even though I think the alignments for what I checked looked OK. So, if I am correctly understanding that the goal is to sequence ~20x (with small fragments such that the genomic coverage can be closer to 15x), then a 2nd set of raw data would get me closer to the 20x-30x that I think is more commonly considered "regular" coverage for inherited/germline variants.
When I received the PDF reports, I noticed something important that I brought to the attention of Basepaws. After a lengthy (but timely) discussion, it was agreed that something was incorrect in the report. My understanding is that this has been reported by at least one other customer, but Basepaws previously not realize the problem with the variant calling strategy (where I believe the earlier emphasis was on trying understand unknown biology, instead of the discussion's shifted emphasis on troubleshooting presentation of earlier findings).
So, due to the fact that this currently affects at least some other customers, I thought I would try to but a blog post together relative earlier than originally planned (with a shift in the intended emphasis, for now).
Reports / Data
1st Sample (Collected 3/14/2019):
Report (as of 6/15/2022): PDF
Report (as of 8/28/2022): PDF (includes corrected "C>T" variant)
FASTQ Read #1: HCWGS0003.23.HCWGS0003_1.fastq.gz
FASTQ Read #2: HCWGS0003.23.HCWGS0003_2.fastq.gz
2nd Sample (Collected 4/21/2022):
Report (as of 6/13/2022): PDF
Report (as of 8/28/2022): PDF (includes corrected "C>T" variant)
Interleaved FASTQ #1 (Run 186, Lane 1): AB.CN.45.31211051000777.LP.858.D9.L1.R186.WGS.fastq.gz
Interleaved FASTQ #2 (Run 186, Lane 2): AB.CN.45.31211051000777.SP.319.D1.L2.R186.WGS.fastq.gz
Interleaved FASTQ #3 (Run 195, Lane 2): AB.CN.45.31211051000777.SP.329.E1.L2.R195.WGS.fastq.gz
As mentioned on GitHub, I will most likely work on writing custom code to create reads in a similar file format as for the 1st WGS sample (which I prefer). At that point, I will add additional links, but the originally provided data is still important.
Incorrect Colorpoint Mutation in Certain v4.0 Reports
(Basepaws Has Corrected Bastu's Reports, as of 8/28/2022)
This is what I thought was important to communicate sooner rather than later.
I was not expecting this to be something to pop-up, and I will try to explain some of the details in a bit. The difference between the report and the data is important, but the issue can also be identified without that.
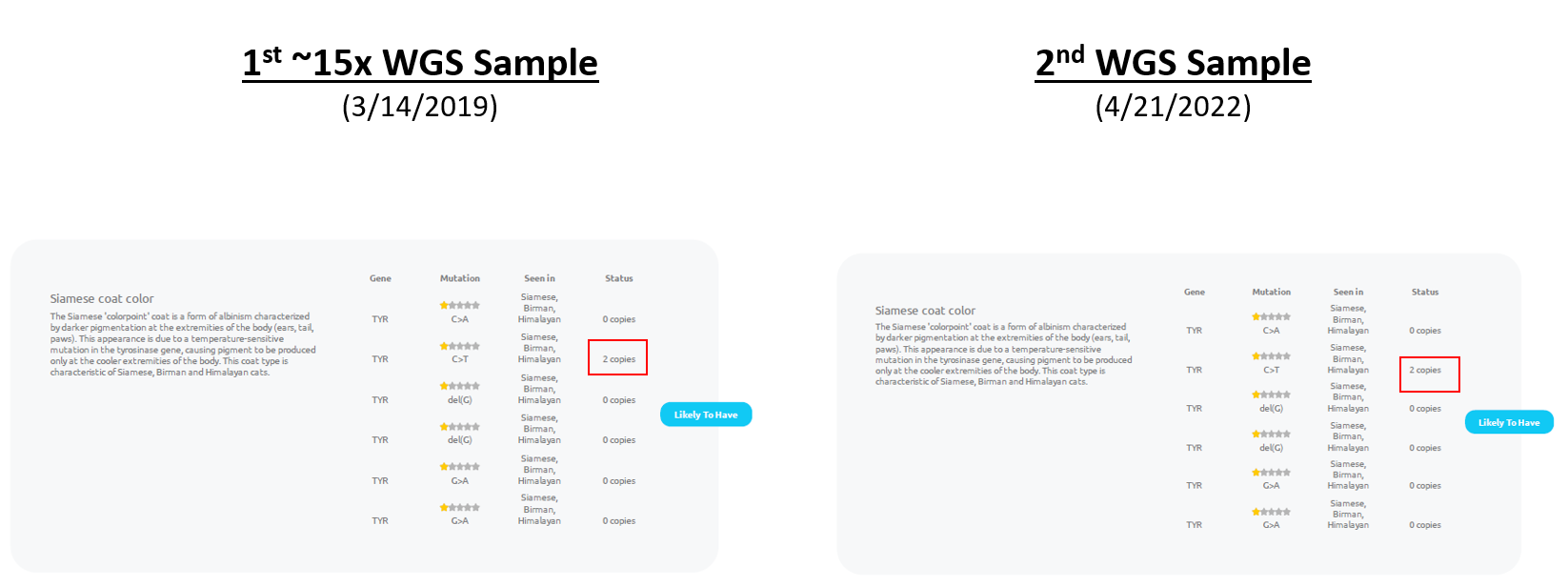
First, my cat was (incorrectly) listed has being likely to have the colorpoint trait in both reports:
As of 8/28/2022, the ability to tell that a report was more difficult than I expected because both sets of screenshots below are labeled as being version 4.0 reports:
Nevertheless, even if looking for the presence of the "black coat color result is not sufficient to tell if the "C>T" colorpoint variant has been corrected, I hope the content of this blog post can help you determine if a given v4.0 is likely to already have the correction or not.
I contacted Basepaws for the following reasons:
- Even though you can have a Tortie Point cat, my cat does not have the colorpoint trait.
- I have submitted samples to essentially all cat genomics organizations / companies that I was aware of. While the markers could theoretically be different, the other results indicated that she has 0 copies of colorpoint mutations. So, I wanted to check if the same variant was being tested.
First, here is a photo of my cat (Bastu):
So, I think is reasonable to say she does
not have the
colorpoint trait. There is also an additional illustration
here (from
this page) that I also think helps explain the colorpoint inheritance pattern.
There is also some discussion regarding traits in
this Basepaws blog post, although I prefer the explanation for
this page from UC-Davis (even though the result that I will show below are for the "
Cat Ancestry" rather than the separate colorpoint test).
The "C/C" result indicates that Bastu should neither have the colorpoint trait nor be a colorpoint carrier.
In terms of Wisdom Panel, you can see the results for some colorpoint markers below:
Bastu's Wisdom Panel results are split among multiple PDF exports of webpages. However, the other traits that can be viewed in the same part of the interface can be seen
here.
After discussion with Basepaws, my understanding is that the specific
C>T mutation is a well-characterized variant, also called the "Siamese" colorpoint variant (with Basepaws staff passing along the
Lyons et al. 2005 publication via e-mail).
There are some additional details from using raw data
here, including selection of
felCat9 chromosomal location to use.
However, after those additional troubleshooting discussions, the alignment to the best matched region of the felCat9 reference genome (with data from my cat's first WGS sample) is shown below:
As a matter of personal preference, I think alignments similar to shown above most clearly show the lack of colorpoint variants (from this position). However, you can also see that from the Amplicon-Seq data that was used for the report (even though these are Whole Genome Sequencing kits). As a special exception, Basepaws provided a file that they also used to say the future reports should report 0 copies instead of 2 copies.
I have a version of that file imported into
Excel with highlighted
100% sequence matches from the
Amplicon-Seq data that I uploaded
here.
I later realized that there is a
1 bp difference for the felCat9 for the amplicon (the "
T" at the end should have been a "C"). The strand of the amplicon is the opposite of the reported nucleotide, but the felCat9 alignment is on the positive strand for the amplicon. In contrast, the amplicon aligns on the negative strand with respect to
felCat8, but the difference is still at the end of the amplicon (which appears at the beginning of the alignment).
So, now, using the raw data, you can tell that the results in future Basepaws reports will need to be changed to reflect that my cat has 0 copies (instead of 2 copies) of the Colorpoint mutation. The lack of mutations now matches her fur color pattern!
Of course, if you also have discrepancies between results from different sources, then you should also contact everybody who is relevant.
If this is a
health marker (instead of a
trait marker) and you can confirm the cause of the discordance in either direction for the same variant (either a false positive or a false negative), then I would also recommend that yous end an e-mail to the
Center for Veterinary Medicine at the FDA. I believe that some technical knowledge is needed to upload data and it is most commonly used for human data, but I have confirmed that veterinary data for pets is allowed on
precisionFDA. Additional effort would be needed to configure an app for cat re-analysis, but all re-analysis of raw data is free through the precisionFDA interface. So, some parts are arguably not ideal for the average consumer, but my point is that a framework to share such data exists and having access to
raw Whole Genome Sequencing data was important for resolving the troubleshooting described above.
Health and Trait Variant Calls
(for current Whole Genome Sequencing Samples)
In my opinion, I believe that the availability of raw data is a unique advantage to Basepaws (at least if you order the Whole Genome Sequencing kit). The process of returning that raw data took some follow-up on my part, but I don't know how many customers intentionally order the kit to be able to receive raw data. I also preferred the fastq.gz formatting for the 1st WGS sample over the 2nd WGS sample, but I am not sure if that might further change in the future.
So, it was to my surprise, the Whole Genome Sequencing data was currently not being used to call the colorpoint variant for my cat.
My understanding is that is also true for the other health and trait markers currently provided. As far as I know, the Amplicon-Seq results are usually OK. However, I hope that either changes or the use of different data types is more clear in the report (even for the "Whole Genome Sequencing" kit), because I was expecting variant calls from the Whole Genome Sequencing data.
There was a problem with using the Amplicon-Seq data for the colorpoint mutation, but my understanding is that programming problems to interpret the raw data were a factor.
Even though Basepaws went back and generated a new Amplicon-Seq library from leftover genomic DNA for my 1st sample (before the Trait markers were offered), I will not be provided the raw Amplicon-Seq data for either sample. My understanding is that this is due to the multiplex design that could be determined from the FASTQ files. I don't think this is ideal. However, as long as the data types are not mixed in one library, I am OK as long as I do receive the raw WGS data. Again, if anything, I would argue the Whole Genome Sequencing (WGS) data appears to be the preferable way to at least evaluate the C>T "Siamese" colorpoint mutation.
Ancestry Results
(uses Whole Genome Sequencing Data)
You can see the results from the 2 Whole Genome Sequencing samples below:
There are some differences, but I think this is starting to get relatively closer to the limitations of my human results. For example, I think my cat mostly has no Exotic purebred relatives (and 1 of 2 reports for the same cat have a 0% Exotic estimate).
However, you can definitely tell that the higher coverage data helps.
Here are earlier plots comparing the low coverage Whole Genome Sequencing (lcWGS) results to my 1st Whole Genome Sequencing result:
"Confident":
"Possible":
You can clearly see additional problems in the ancestry results for the less expensive kits, with much lower coverage data used to impute genotypes.
I am not sure how much the relative revision of genomic reference sequence for the cat versus human may also matter, but I would guess that might have some impact on making a fraction of the genotypes harder to impute (and, therefore, the ancestry harder to predict).
This also gives me the impression that the new reports may only be providing the "possible" results (without the option to view the "confident" results that I prefer). However, I will follow-up with Basepaws on that. Either way, I would say the higher coverage data helped, but I think that is most clear with the "possible" confidence results.
Nevertheless, even at whatever is used as the current setting, you can see my cat has mostly Western and Polycat ancestry. That is consistent with what I see from other companies and organizations for the same cat.
Eukaryotic Metagenomic Results
(can vary over time)
You can see the results from the 2 Whole Genome Sequencing samples below:
I am not currently sure what to think about this.
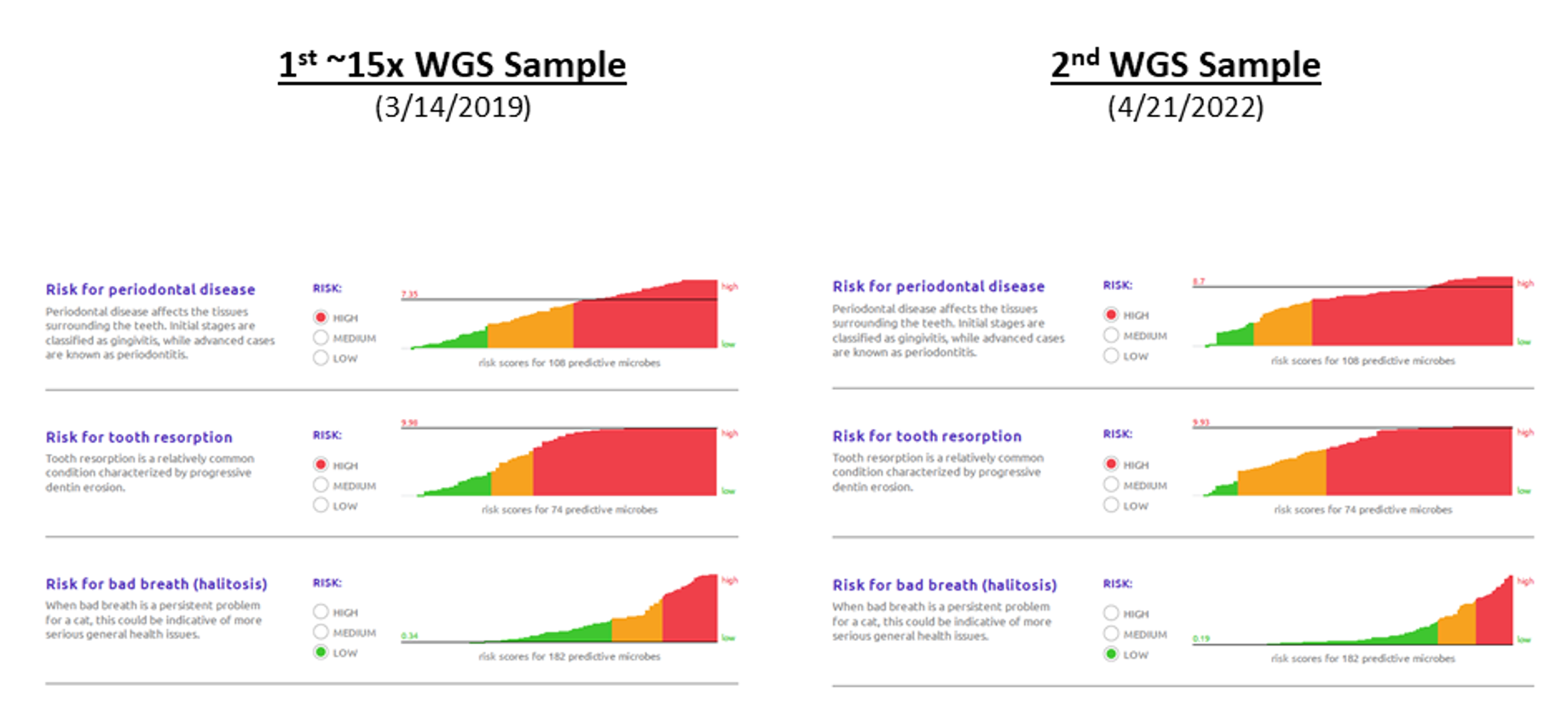
Dental Health Scores
(can vary over time)
You can see the results from the 2 Whole Genome Sequencing samples below:
This is something that I hope that I can develop more of an opinion about over the next year or so. For example, I have started a
GitHub discussion where I list some things that I would like to look into and I certainly welcome additional feedback.
Similarly, I have already started a subfolder on this topic
on GitHub that contains data/results and my thoughts so far.
One one hand, Basepaws has increased by general awareness of dental health in my cat. For example, I have started discussions with my vet, and I think that is good.
However, I do not yet have a complete opinion regarding the Dental Health Test results. For example, I would not actively recommend the Dental Health Test, but I am also not currently confident that there is a problem with the results that I can clearly explain.
Nevertheless, there are a few things that I would like to point out or comment about:
1) As I understand it, I think my cat has some of the highest "High Risk" scores that are plotted. However, after multiple discussions with the vet, my understanding is that I should describe my cat as being in very good dental health for a cat her age.
My cat has been started on prescription dental diet, although my understanding is that is primarily a preventative measure given her age (and possibly my questions about dental health). I will also take my cat in for the recommended yearly cleanings, and I believe that the vet confirmed that she doesn't currently need to come in more often.
Also, I believe that Basepaws has confirmed in an e-mail that the goal is to estimate risk and not to provide a diagnosis. In other words, you can have a cat with a serious problem with low risk in the Basepaws Dental Health Test, and you can have a cat without a serious problem with high estimated risk. My understanding is that cat falls in the second category. The point is that something else needs to be performed by the vet to determine if your cat has a problem.
2) I saw an e-mail from 6/8/2022 titled "
See what saved this kitty's life..." with a link to
a blog post (which I believe was posted on 7/12/2021).
My initial reading and what I understood with re-reading were different. Because the test was collected considerably before any problems were observed, I think that is less of an concern.
However, what could have concerned me is if somebody noticed their cat had bad breath, ordered a Basepaws Dental Health Test, and then went to see the vet after receiving results from Basepaws 6 weeks later.
In other words, theoretically, I think this could have increased the risk of harm if there was an urgent problem. The reason is that the Basepaws Dental Health Test can't tell you if your cat has a problem or not, and I would say you should start the process of trying to see your vet as soon as you notice a problem (not after waiting for a results that make take 6 weeks or more). I could imagine a vet wanting to see if a symptom like bad breath persisted for some time before suggesting scheduling an appointment to bring the cat into the vet. However, I would guess this waiting time might be more on the scale of days instead of more than a month?
For example, my understanding is the other cat was at "medium" risk in all 3 categories, which I would interpret as meaning the cat was at lower risk for than my cat (2 "high risk" scores and 1 "low risk" score for bad breath). However, my cat had no serious dental health problems, and this cat with what I would consider to be at lower risk had a serious dental health problem.
I also had a question of "medium" risk versus "average" risk, but I have moved that to a footnote1.
So, I would at least consider bad breath a reason to call the vet and ask if the cat should come in. I would not order a Basepaws Dental Health Test because my cat has bad breath (or any other novel symptom causing me concerns), and then wait for the results before scheduling an appointment with the vet. I have also confirmed that Basepaws agrees you should not delay seeing your vet in order to obtain a Basepaws Dental Health Test result when you see a worrying symptom.
To be fair, I believe that I have seen at least one cat diagnosed to have a dental health problem with 3 "high risk" scores on the
Facebook Basepaws Cat Club. So, I hope that is helpful in getting an overall sense of the results. However, I think the above examples are also important to take into consideration.
3) I think a well-characterized disease variant that has a second validation from the clinic could be different. However, for the reasons above, I would have preferred that the Dental Health Risk scores were not added to my cat's medical record because of the discussion most immediately above.
However, I think the local vets have started to get to know my cat as an individual patient, so I would guess this is probably not an issue unless I had to see another vet (and that vet gave too much emphasis to those scores).
4) I remember previously reading about critiques from professors of veterinary medicine regarding the Basepaws Dental Health test in
this Los Angeles Times article. I have also added some comments on the
preprint for the Basepaws Dental Health test, and I hope that others with relevant experience will do so as well (and I hope that there can be responses to comments from others).
However, again, I would certainly like to learn more! You can either provide feedback as a comment, or there is a "
discussion" enabled on the GitHub page.
Footnotes:
1When I asked about what represented "
average" risk, Basepaws provided a link to the Dental Health Test
whitepaper. This is similar but not identical to the
preprint. So, I don't have a direct answer. However, I can say that the preprint and the tables
here were helpful in having discussions for my vet.
For example, I added an
additional comment to the preprint regarding the
positive predictive value estimation after talking to the vet.
The numbers in the whitepaper are slightly different (for example, there are 441 periodontal disease cats in the whitepaper and 570 periodontal disease cats in the preprint). Nevertheless, if I use the preprint numbers, then I had possible estimates of positive predictive values for the combined high/medium group where I estimated the periodontal disease score might have positive predictive values of roughly 50% and the tooth resorption score might have positive predictive values of roughly 20%.
In every calculation that I attempt, the positive predictive value is noticeably lower for tooth resorption than periodontal disease.
For periodontal disease, I think the prevalence estimates and stage of the disease are important, but I wonder if might help to pose the question of whether something different than the standard recommended care should take place. My current thought is to use the the vet's individual assessment (as independently from any estimate as possible) over the risk estimate, but I think conversations with your vet are worthwhile. Basepaws clearly indicates the risk estimates should not be used as a diagnostic for dental health.
Change Log:
7/2/2022 - public post date.
7/3/2022 - add link to Dental Health Test GitHub discussion; also, as I went back to the possible confusion matrices that I created from the preprint to attempt to estimate the positive predictive value, I decided change wording for describing "medium" risk versus "average" risk as I look into the topic more; minor changes. 7/4/2022 - minor corrections; revise description of timing for other cat's dental health test.
7/7/2022 - update post after receiving e-mail response from Basepaws
7/10/2022 - add date to separate Basepaws e-mail from Basepaws blog post
7/16/2022 - formatting changes
7/17/2022 - after looking at 59 posted Dental Health Test reports, switch to only using the higher positive predictive value estimate. The score distribution is different than the true prevalence, but that made me feel a little better about the possible positive predictive value.
7/20/2022 - minor changes + change footnote content
7/23/2022 - add additional UC-Davis VGL colorpoint link + minor formatting changes; add description of BLAT result with a minor difference from what I originally thought (mismatch at end, but not within BLAT hit); add link to product towards the beginning of the blog post.
7/24/2022 - re-arrange colorpoint background and provide photo of Bastu.
7/31/2022 - minor change to make Amplicon-Seq link earlier to find.
8/8/2022 - add link to Basepaws blog post acknowledging the fix.
8/14/2022 - add links to raw data for 2nd WGS sample (in format provided by Basepaws); update/modify section related to raw WGS data return and use of WGS data in reports.
8/16/2022 - formatting changes to make 2 hyperlinks easier to find.
8/23/2022 - upload alternative screenshot for Basepaws Siamese Coat Color result that is incorrect in the v4.0 report.
8/27/2022 - minor formatting changes
8/28/2022 - change tense for Basepaws correction, but also add content to explain how I have different v4.0 reports downloaded at different dates that are both before and after the "C>T" colorpoint variant correction; also, add links to reports that are downloaded more recently (with the corrected variant result).